ImmunExplorer (IMEX) is a software for analyzing IMGT/HighV-QUEST output files and enables a wide range of statistical analyses of clonality and diversity in NGS IG and TR data.
Introduction
ImmunExplorer (IMEX) uses preprocessed next-generation high-throughput and deep sequencing (NGS) data from human immunoglobulins or antibodies (IG) and
T cell receptors (TR) derived from the international ImMunoGeneTics (IMGT®) information system® .
IMEX is a user-friendly framework for performing descriptive statistic analyses, clonal expansion and diversity experiments based on the
CDR3 sequences and V-(D)-J elements (nucleotide, amino acid level), primer efficiency analyses, and
various different visualization experiments based on IMGT® compressed output files.
The software is developed by researchers of the Bioinformatics Research Group at FH OÖ, Hagenberg Campus and researchers of the Immunogenetics Department at the Red Cross Transfusion Center in Linz.
Requirements and Installation of IMEX
IMEX Windows Only:
System requirements:- IMEX is available for Windows operating systems (64-bit)
- Installed memory (RAM): minimum 16 GB
- Microsoft .NET 4.0
- Internet access for downloading gene lists derived from the IMGT® information system.
- Carefully read the license agreement and proceed only if you agree to the terms and conditions.
- Please download the stable version here (as .zip file):
- ImmunExplorer v. 1.1
- Additional data for testing IMEX can be downloaded here: small data sets (without files for testing the splitter and merger) and big data sets (includes data for the splitter and merger).
- If there are problems with firewall settings or if internet access is limited, please download the gene lists for IG and TR here. Copy the gene lists in the current folder. The current folder is located default in C:/public/imex/current/.
- Right click on the downloaded *.zip file and select the menu item "Properties" in the context menu.
- If this property is visible, click "Unblock" and "OK" to close the "Properties" window.
- Extract the content of the downloaded .zip file in a separate folder.
- Navigate to the extracted folder and double-click on ImmunExplorer.exe.
IMEX Command-line for Windows/Linux/Mac OS X
Please download the latest version for Windows/Linux/Mac OS X: ImmunExplorer v.1.0 betaSetup and Introduction:
- Please download the current gene list (imgtGenesFile in settings.xml) provided by the IMGT® information system of the IMGT/GENE-DB here.
-
Open settings.xml in a text editor and adapt or modify all paths of the following tags:
- imgtGenesFile
- primerTRFile
- primerIGFile
- directory
- path
- Be sure that the primer files and the gene file (downloaded from the IMGT® GENE-DB) are in the directory that in the settings.xml file has been defined
- Start statistical and comparison analysis of IMEX by running (mono) ImmunExplorer.exe myIMGTdataFOLDER settings.xml -gl. A folder "genes" in the myIMGTdataFOLDER should be available now.
- Using (mono) ImmunExplorer.exe -h or (mono) ImmunExplorer -help shows all arguments:
- -gl: creates gene and allele files for V-(D)-J comparison analyses
- -split: splits FASTA files
- -merge: merges preprocessed IMGT/HighV-QUEST output files
- -basics: performs basic statistical analysis
- -clonality: calculated clonotype frequencies of the specified gene/sequences and determined CDR aa length distribution frequencies
- -diversity: calculates diversity and estimates the diversity by using a mathematical model and parameter optimization
- -primer: performs primer analysis
- -vdj: calculates multiple gene/allele V-(D)-J gene arrangements
- -compare: enables comparison experiments of two or multiple IMGT/HighV-QUEST output files of the same receptor type
Usage:
Windows:
- Extract the downloaded .zip file
- Navigate to the extracted ImmunExplorer folder in a command-line user interface (Windows Command Prompt, e.g.)
- Start statistical and comparison analyses of ImmunExplorer by running "ImmunExplorer.exe myIMGTdataFOLDER|myFastaFile.fa|myIMGTFile.zip settings.xml -analysisType".
Linux/Mac OS X
- The mono runtime environment version 4.0.1 or higher have to be installed. Some Linux distributions already come with an installed mono. To check which version you have installed please run "mono -V" on a terminal.
- Mono can be installed by using "aptitude install mono" (Linux) or please follow the installation instructions for Linux or for Mac OS X.
- Extract the ImmunExplorer archive and navigate to the extracted folder in a terminal.
- Please modify the settings.xml file as described in the Usage section. (Please use correct paths!)
- Run statistical analyses and comparison experiments of ImmunExplorer by using "mono ImmunExplorer.exe myIMGT.zip|myFASTA.fa|myIMGTdataFOLDER settings.xml -analysisType"
How to use IMEX
After starting ImmunExplorer.exe, a startpage offering the following algorithms is shown:
- Preprocessing methods for uploading the files on the IMGT/HighV-QUEST information system
- Postprocessing methods for merging the output files derived from the IMGT/HighV-QUEST (if the files have more than 500,000 sequences included)
- Single and multi IMGT/HighV-QUEST output file analyses
- Comparison analyses of IMGT/HighV-QUEST output files
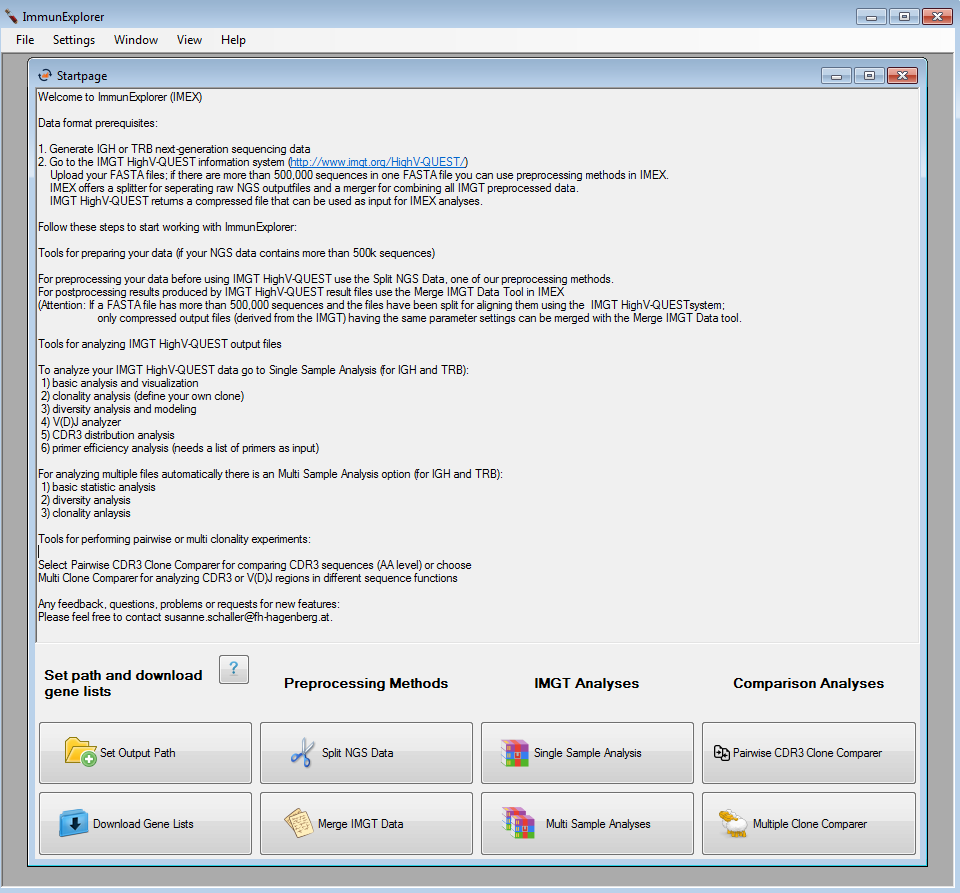
|
|
Figure 1: Startscreen of IMEX. |
Configuration Settings
Two settings have to be configured before IMGT® analyses can be performed properly:
-
First, an output path has to be set. This is the output path where normally (and by default) all result files are saved.
-
Second, gene lists from the IMGT® information system have to be downloaded for V-(D)-J plots and V-(D)-J analysis. As we currently allow gene analyses only for Homo Sapiens and for the beta chain of the IG and TR only those gene lists can be downloaded (and allelic information). You can find the downloaded gene lists at the pre-defined path location in the current folder.
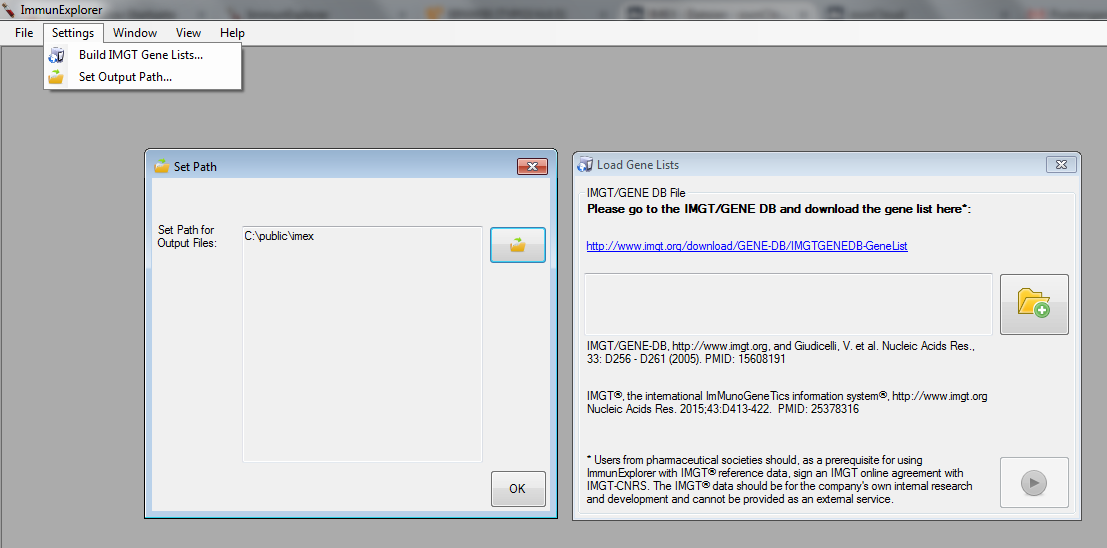
|
|
Figure 2: Path configuration window (left) and gene list update window (right). |
Preprocessing Methods
The preprocessing methods provided in IMEX enable splitting FASTA (or text) files: Currently the IMGT/HighV-QUEST information system enables the upload of FASTA files containing max. 500,000 sequences. If your file contains more sequences use the Split NGS Data tool of IMEX and prepare the data for the IMGT upload. (Of course you can use the splitter also for FASTA files with less than 500k sequences for your own preprocessing purposes.) After preprocessing the files using IMGT/HighV-QUEST and splitting the original FASTA files, you can use the file merger offered by IMEX. It is important that for all merged parts were created using the same parameter settings having the same filename including the ending "_PART_X.zip" (e.g., PatientX890_PART_1.zip and PatientX890_PART_2.zip and so on).
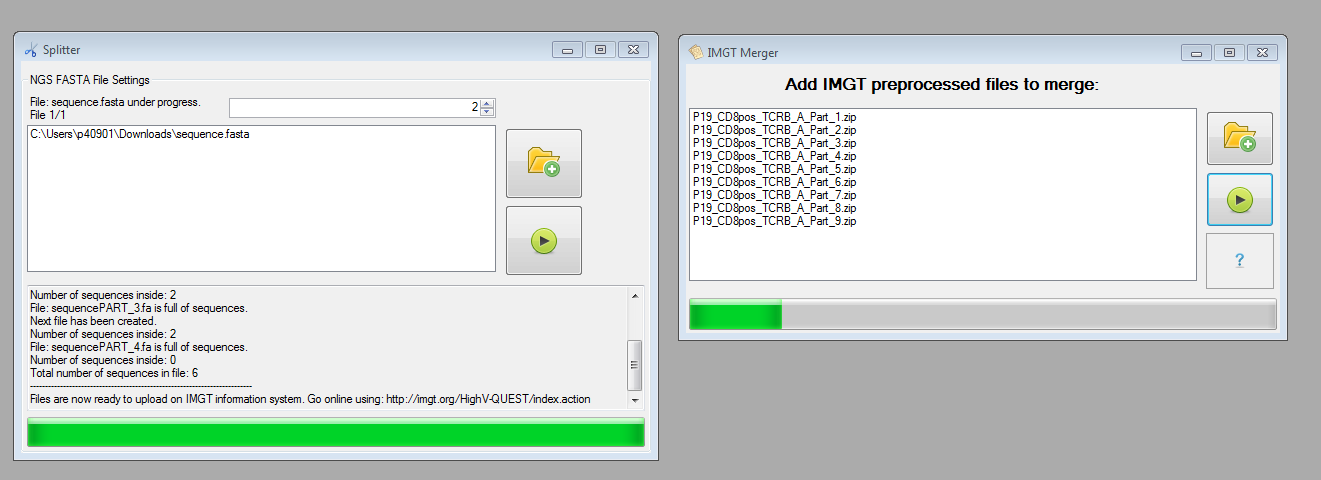
|
|
Figure 3: Preprocessing methods: splitter and merger.
|
Sample Analyses
Single Sample Analysis
You can now load IMGT/HighV-QUEST output files into IMEX and use the following analysis methods:
- Descriptive statistic analyzer and visualizer
- Clone analyzer and visualizer
- Primer analyzer and visualizer
- V-(D)-J analyzer and visualizer
- Diversity analyzer and parameter optimization
- Clone tracker and machine learning (to be released soon hopefully)
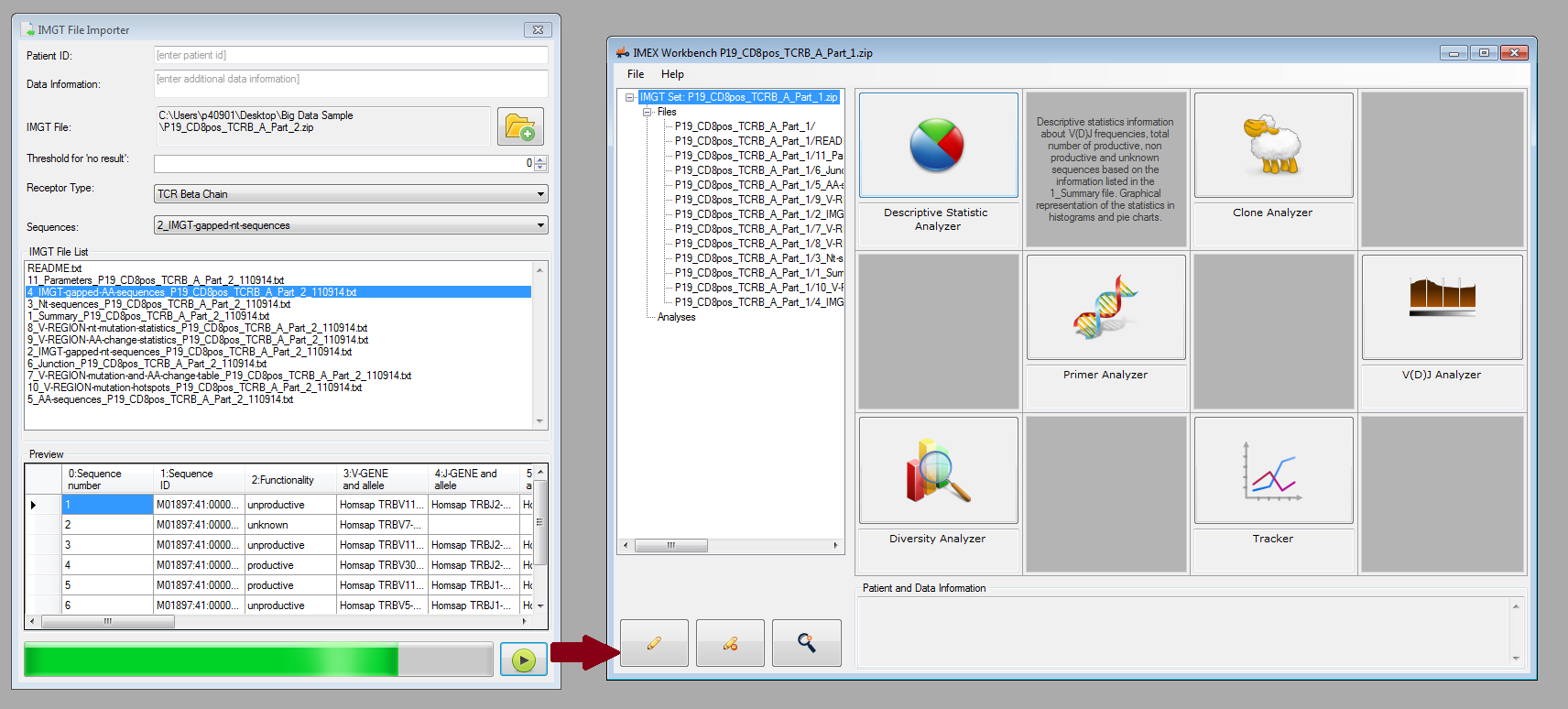
|
|
Figure 4: Single sample analysis: file importer (left) and main workbench for choosing different analysis types.
|
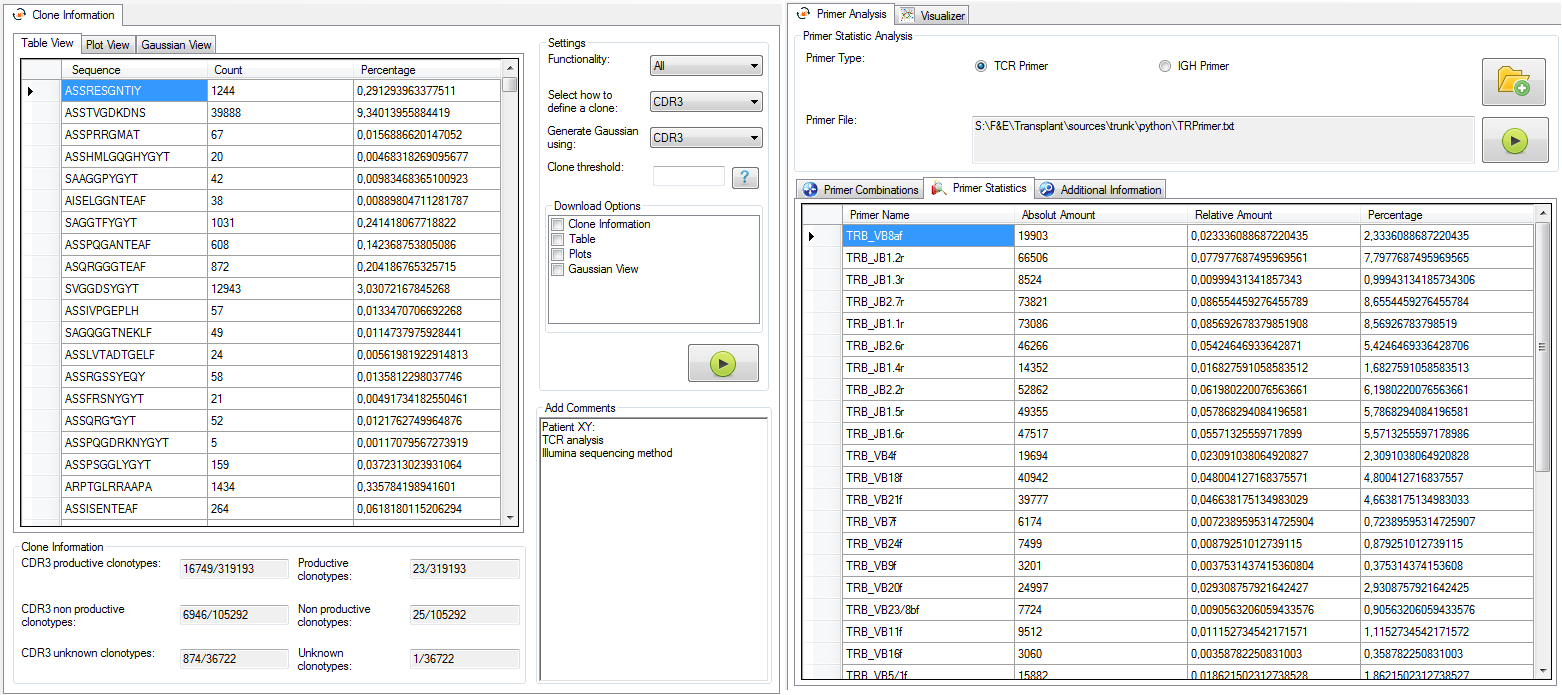
|
|
Figure 5: Clone view (left) and primer view (right). |
Multi Sample Analysis
IMEX offers several methods for analysing and comparing samples. By now, descriptive statistics, clone analysis based on the CDR3 sequences, and diversity analysis can be performed automatically. The output files are located on the path you have selected at the beginning.
Comparison Analyses
IMEX offers the following clone comparison methods:- Pairwise clone comparer
- Multi clone comparer
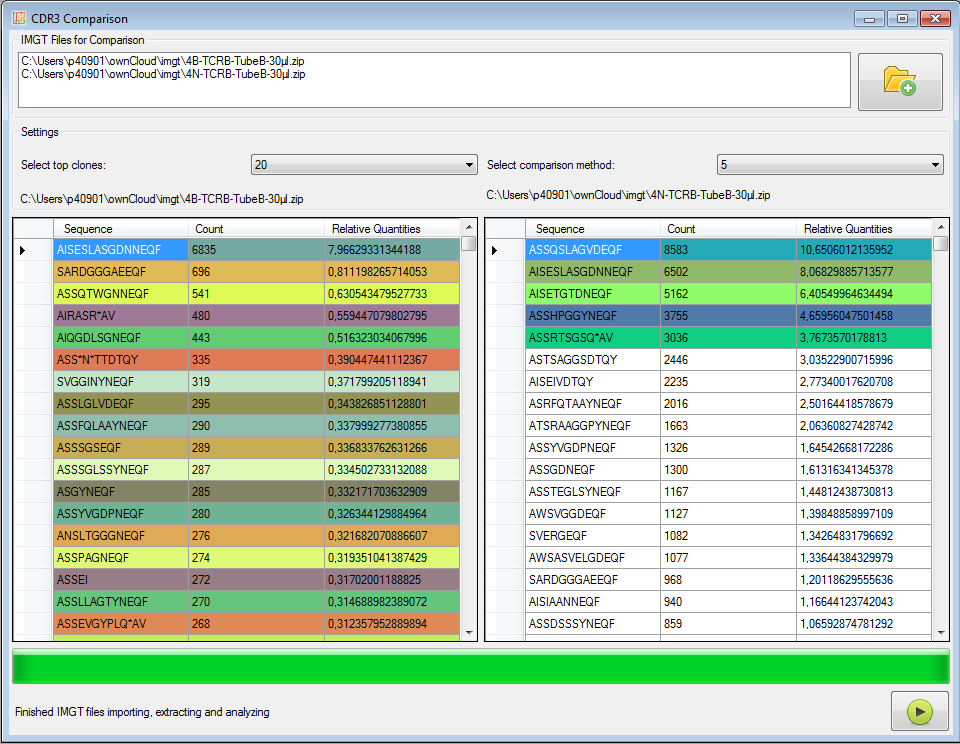
|
Figure 6: Pairwise clone comparer (CDR3 sequences, AA level).
In each input file the top x clones are collected and their frequencies are calculated also for the other input file.
|
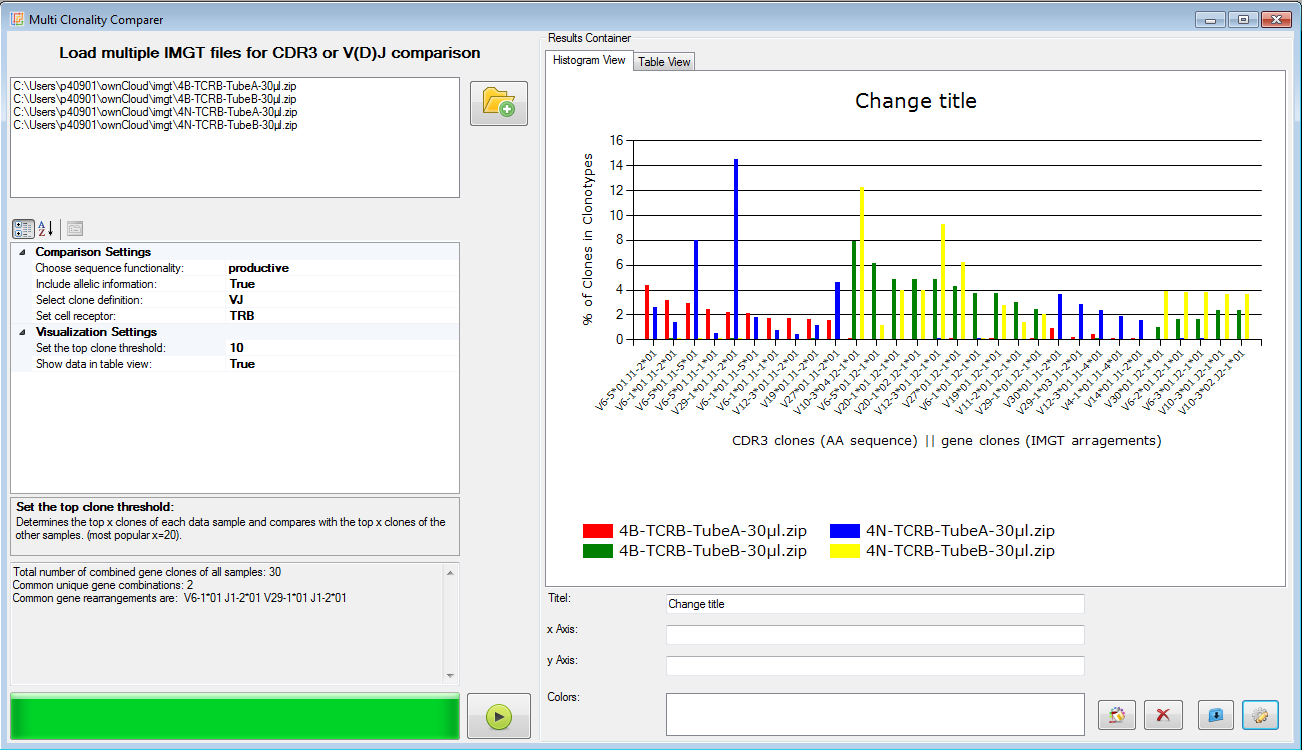
|
|
Figure 7: Multi clone comparer (here shown for V-J clones). |
All icons used in the software IMEX and icons that are visible on the screenshots are downloaded from IconArchive .
Contact
This research project is a collaboration of the Cross Blood Transfusion Service for Upper Austria, Linz and the Bioinformatics Research Group at the University of Applied Sciences Upper Austria, Hagenberg Campus. If there are any questions, problems, ideas please do not hesitate to contact Susanne Schaller, Johannes Weinberger, Martin Danzer or Stephan Winkler.
Relevant Publications
-
S. Schaller, J. Weinberger, M. Danzer, S. M. Winkler: Detailed Analysis of Clonality, Diversity, and Gene Frequency Distributions in B- and T-Cell Receptors based on NGS Data. Revolutionizing Next-Generation Sequencing: Tools and Technologies, Leuven, Belgium, 2015.
-
S. Schaller, J. Weinberger, M. Danzer, S. M. Winkler: Modelling States of Human Adaptive Immune Systems by Analyzing B- and T-Cell Receptors using Machine Learning. Proceedings of the Computer Aided Systems Theory EUROCAST 2015, Las Palmas de Gran Canaria, Spanien, 2015.
-
J. Weinberger, R. Jimenez-Heredia, S. Schaller, S. Suessner, J. Sunzenauer, R. Reindl-Schwaighofer, S. Winkler, C. Gabriel, M. Danzer, R. Oberbauer: Immune repertoire profiling reveals that clonally expanded B- and T-cells infiltrating diseased human kidneys can be tracked in blood [to be published 2015].
-
S. Schaller, J. Weinberger, M. Danzer, S. M. Winkler: ImmunExplorer: A Framework for NGS-based Characterization and Visualization of the Human Immune System. Proceedings of the 22nd Annual International Conference on Intelligent Systems for Molecular Biology (ISMB), Boston, MA, USA, 2014.
-
S. Schaller, J. Weinberger, M. Danzer, C. Gabriel, R. Oberbauer, S. M. Winkler: Mathematical Modeling of the Diversity in Human B and T Cell Receptors using Machine Learning. Proceedings of the 26th European Modeling and Simulation Symposium EMSS 2014, Bordeaux, France, 2014.